

Yves here. It’s easy to be cynical about the state of medical and biomedical science, given how the Biden Administration set out to trash its brand. Invocations of “Follow the science” as the justification for vaccine mandates that could not stop transmission of Covid, particularly after variants like Omicron had mutated to be markedly different and therefore substantially evasive of vaccines based on the “wild type” virus. Even worse, repeat boosting with mRNA vaccines produces only a short-term boost in immunity and negative efficacy in the intermediate term. The gap between the aggressive and insistent messaging and results over time have produced a backlash against vaccines generally. Well done, Team Biden!
Another reason for layperson leeriness is (at least in the US), aggressive hyping on TV of expensive medications. Those ads have the effect of reminding patients of how much health care, American style, has come to be about profits, not medical outcomes.
So KLG provides an important corrective below, of two important advances, both in treating dangerous genetic diseases.
By KLG, who has held research and academic positions in three US medical schools since 1995 and is currently Professor of Biochemistry and Associate Dean. He has performed and directed research on protein structure, function, and evolution; cell adhesion and motility; the mechanism of viral fusion proteins; and assembly of the vertebrate heart. He has served on national review panels of both public and private funding agencies, and his research and that of his students has been funded by the American Heart Association, American Cancer Society, and National Institutes of Health
Sometimes it can be forgotten that beyond the hype, modern biomedical science can be a wondrous thing. Here are two examples of the why and the how, with some of the relevant history attached, from recent developments in the treatment of two common, debilitating genetic diseases of the blood, one sex-linked and the other not: hemophilia and sickle cell disease.
Hemophilia. Hemophilia A (HA) and Hemophilia B (HB) are distinct genetic diseases that are clinically indistinguishable. HA is caused by a deficiency of Factor VIII (F8) in the blood coagulation cascade responsible for hemostasis, while HB is caused by a lack of Factor IX (F9). Hemostasis is a constant challenge. This video (3:21) from the National Hemophilia Foundation is a remarkable visual explanation of the clotting cascade, provided you ignore the necessary simplification of the process.
HA and HB are both X-linked recessive disease [1], meaning that the genes for F8 and F9 are on the X chromosome and males have the disease with they get the X chromosome with the mutant (x). Thus, a normal female will have the genotype XX (both X chromosomes contain a normal copy of F8 or F9), while a phenotypically normal carrier femalewith a normal clotting cascade and no evidence of hemophilia will be Xx (one normal, one mutant F8 or F9). A person with HA or HB will have the genotype xY, with the “wrong” copy inherited from his mother. The most well-known kindred with multiple members with hemophilia is that descended from Queen Victoria [2]. The most famous member of this extended family is undoubtedly Alexei, the youngest child of Nicholas II and Alexandra. It turns out that the Tsarevich had Hemophilia B, caused by F9 deficiency due to an mRNA splicing defect that produces an inactive truncated F9. As with most genetic diseases, multiple mutations have been identified in HA and HB. OMIM [3], what I call “Wikipedia for Smart Medical Students,” is essential when considering any and all human genetic diseases. Sometimes too much information, though, and I stopped counting the number of mutants identified in HA.
HA and HB patients form blood clots very slowly due to their lack of enough F8 and F9 in the circulation. Untreated, severe hemophilia (less than 1% of normal F8 or F9 level) leads to spontaneous and recurrent musculoskeletal, soft tissue, and other life-threatening bleeds, including intracranial hemorrhage. Bleeding following trauma or surgery is very difficult to manage in these patients. Repeated bleeding into joints leads to destructive arthropathy and severe disability. Patients with moderate hemophilia have F8/F9 levels at 1-5% of normal. In patients with mild hemophilia, severe bleeding is generally restricted to trauma, but these patients are at greater risk of intracranial bleeding. Any manifestation of hemophilia is serious.
Effective, if onerous treatments (e.g., infusion of F8 or F9 every few days), for HA and HB, respectively, have been available since the 1950s (Figure 1).
Before these interventions hemophilia was treated no more effectively than the incantations of Rasputin in the final royal court of Russia. These treatments began with the infusion of fresh frozen plasma (FFP) in the 1950s and later plasma cryoprecipitate (precipitated residue collected from thawed FFP) or lyophilized (freeze-dried) plasma. These preparations contained enough F8 or F9 for normal blood clotting but circulating levels of the exogenous clotting factor declined rapidly, necessitating frequent treatment. Just as critical, there was no way to test for or remove viruses from these preparations, and hemophiliacs receiving these products the 1970s and 1980s were severely at-risk for viral hepatitis, e.g., Hepatitis C, and HIV. During this fraught time Ryan White (1971-1990) of Kokomo, Indiana, became one of the earliest human faces of the HIV/AIDS epidemic.
The initial development of recombinant DNA and gene cloning in the early-1980’s presaged better therapies for hemophilia and other genetic diseases [4]. The gene for F9/HB was cloned in 1982 and F8/HA in 1984. Recombinant F8 (i.e., cloned F8) for treatment of HA patients became available five years later in 1989. The first demonstration of successful gene therapy for HB was in 2011, 29 years after the gene was cloned. The same for HA was demonstrated in 2015 (31 years after the gene was cloned). Clinical trials for HA and HB have now been completed and effective gene therapy for these devastating diseases is a real possibility.
Why did this take so long? Recombinant DNA is “easy” today, but it was not 40 years ago when each molecular biology laboratory worked almost from scratch on every project [5]. Gene therapy also requires that the cloned therapeutic gene be delivered effectively and expressed appropriately in the proper target tissue. Mammalian genes are generally not simply turned off and on as they are in bacteria such as Escherichia coli (E. coli), which was from the beginning and remains the workhorse of molecular biology. Unmodified viruses that infect humans are an answer to both conundrums, but unmodified viruses “live” by taking over their target cells and making more viruses. Sometimes this is benign, sometimes not. By definition, great care must be taken.
Research on adenoviruses (AV) and adeno-associated viruses (AAV) provided the path that led to gene therapy for hemophilia. These genetic vectors (stripped down genetic elements that deliver recombinant genes to the target tissue where they are expressed) have long been used to express exogenous proteins in cultured cells and other experimental models where the stakes are low. The path to human gene therapy has been difficult, as illustrated by the story of Jesse Gelsinger (1981-1999) who died in an early gene therapy experiment that went horribly wrong at the University of Pennsylvania, then as now one of the leaders in gene therapy research and development. The case of young Mr. Gelsinger was a much more than sobering lesson to thousands of scientists and clinicians who viewed gene therapy as the “next great thing” in clinical medicine. From the Jesse Gelsinger link above: “In a flash the field of gene therapy collapsed, taking its grandiose promises of miracle cures along with it.” Yes, it did. I was just beginning my first independent academic position, and this was the topic of the day for a long time (and should never be forgotten). But twenty-four years later things are different, and gene therapy again can be a next great thing in biomedicine.
But why hemophilia in particular? The keys for hemophilia are several:
- F8 and F9 are not large proteins, so the DNA sequence encoding their mRNA can “fit into” the vectors used to deliver them to their target tissues. This is a non-trivial problem for experimental and clinical use of AV and AAV vectors.
- AAV-5 has been developed as the vector of choice because it is versatile, efficient, and unlikely to cause off-target pathology at the doses necessary, such as the unexpected immune hyper-response to adenovirus that killed Jesse Gelsinger in 1999.
- The proteins of the coagulation cascade are produced in the liver and secreted into the blood. So, the target tissue for hemophilia is the liver, which is easy to access by simple infusion of the AAV-5 preparation. Liver cells (hepatocytes) are then transduced by AAV-5, and the coagulation factor is produced and secreted into the circulation through the normal pathway. The liver is our second largest organ (second to skin) and is “amenable to manipulation,” for lack of a better concept.
- Replacement of 5-10% of the normal levels of F8/F9 is sufficient to prevent HA and HB. Normal expression levels of ~50% (carrier, Xx) or 100% (normal, XX or XY) for these proteins are not required to cure the disease. Thus, the cell biology of F8/F9 expression and secretion can be relatively inefficient and still be effective.
For those who want to dig deeper into the details, Table 1 here summarizes the relevant clinical trials. These trials are very convincing in my view. As it happens, transduction of liver cells by AAV-5 expressing F8 or F9 produces a response that cures HA/HB for at least five years. Compared to semi-weekly infusions of recombinant F8 or F9, this is a cure that works in the long term. The persistence of the response is likely to be increased as improvements are made in the transduction protocols. But the road has been long, 41 years since the gene for F9 was cloned and 39 years for F8. As I have noted here before, biology and biomedical science are nearly always incremental. Shortcuts are few, and sometimes lead to poor outcomes, clinically and otherwise. Patience and persistence are often rewarded, though, when the means to complete the task remain available. Gene therapy for hemophilia is likely to be a next great clinical advance.
Sickle cell disease. Sir Archibald Garrod identified “inborn errors in metabolism” and showed they are inherited beginning with his work on alkaptonuria near the turn of the 20th century, but sickle cell disease is known as the first genetic disease, properly identified as such. From the beginning of its recognition in the African American population, sickle cell anemia/sickle cell disease was described in its clinical manifestations by a series of physicians who may have practiced “primitive” medicine compared to what we have now. But they were also complete physicians who treated their patients as individuals instead of “conditions,” which is an enduring lesson.
The first description of sickle cell disease, “Peculiar Elongated and Sickle-shaped Red Blood Corpuscles in a Case of Severe Anemia,” was published by Dr. James B. Herrick of Chicago in 1910 in Archives of Internal Medicine, Volume 5, p. 517.
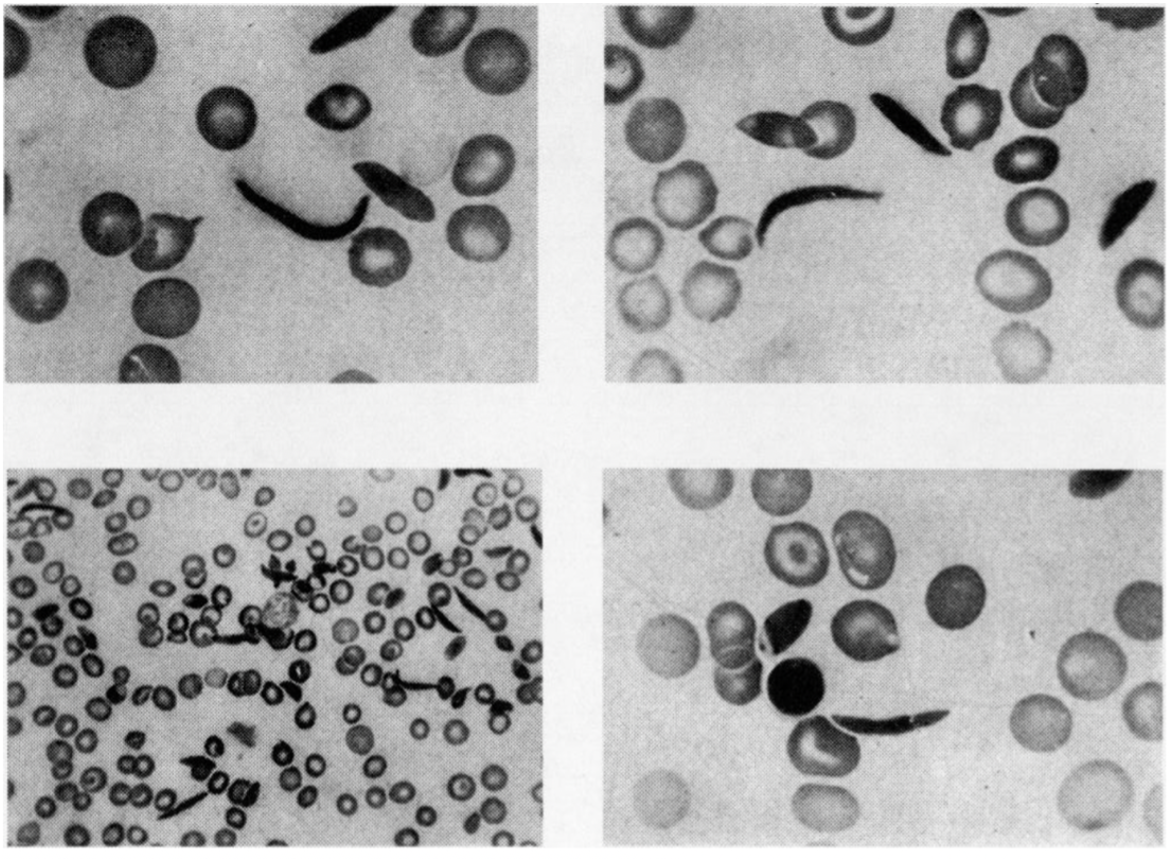
The paper has been reprinted in a more accessible location as a Classic of Biology and Medicine in Yale Journal of Biology and Medicine (pdf, 2001). Herrick’s photomicrographs of sickle cells are just as good as any images produced since (the image here is rotated 90 degrees; the lower magnification is at the lower left). Dr. John G. Huck of Baltimore described the complete symptomology of sickle cell disease in 1923 in a paper published in the Johns Hopkins Hospital Bulletin. His patients were from two large kindreds of African Americans in Baltimore. Based on Huck’s work, W.H. Taliaferro showed that “Sickle-cell anaemia…is an inherited condition and behaves as a single Mendelian character which is dominant over the normal condition and is not sex-linked” (pdf here) [6].
The next advance in the understanding of sickle cell disease came 24 years later in 1947 when Linus Pauling, Harvey A. Itano, S. J. Singer, and Ibert C. Wells published “Sickle Cell Anemia, a Molecular Disease” in Science (paywall, but this recent review is available) [7]. This paper is remarkable for what was accomplished by careful reasoning and even more careful biochemistry when the discipline required more thought than action. Based on clinical observations and genetic studies referred to above, the hypothesis was that “erythrocytes of certain individuals possess the capacity to undergo reversible changes in shape in response to changes in the partial pressure of oxygen,” the latter at high altitudes or under stress. Their primary conclusion was that one amino acid with a negative charge was changed to an amino acid that is neutral in the mutant HbS.
Ten years later, V.M. Ingram (paywall) at the Cavendish Laboratory in Cambridge determined “that out of nearly 300 amino acids in the two proteins (alpha and beta) only one is different; one of the glutamic acid residues of normal haemoglobin is replaced by a valine residue in sickle cell anemia haemoglobin.” Glutamic acid has a negative charge; valine in neutral. That only one amino acid change causes such disorder is another argument against facile conjectures sometimes found in current biomedicine that such a minor change in protein structure, i.e., one different amino acid out of 300, will be inconsequential. As a minor aside with great importance, it is not an accident that three Nobelists are acknowledged in Ingram’s short paper: Sydney Brenner, Francis Crick, and Max Perutz. Cambridge was then and still is the best place to do biochemistry.
In addition to determining that the charge in HbS is different, the Pauling laboratory also noted that this single change in HbS caused the protein to polymerize under low oxygen tension: “Under appropriate conditions, then, the sickle cell anemia hemoglobin molecules might be capable of interacting with one another at these sites sufficiently to cause at least a partial alignment of the molecules withing the cell, resulting in the erythrocytes becoming birefringent [8] and the cell membrane’s being distorted to accommodate the now relatively rigid structures within its confines.”
This is exactly what happens in sickle cell disease. At low oxygen concentration the HbS-containing tetramers polymerize and deform red blood cells. Subsequent damage to the microvasculature leads to frank sickle cell disease and anemia associated with destruction of sickle cells. Thus, the proximal cause of sickle cell disease is polymerization of mutant hemoglobin. It follows that inhibition of HbS polymerization will prevent the disease.
The drug candidate voxelotor was first described about five years ago as a compound that “improved” the properties of sickle cell disease blood under deoxygenated conditions. It was subsequently shown to bind to the alpha-globin chain of hemoglobin and increase hemoglobin oxygen affinity, thereby decreasing the polymerization tendency of deoxygenated HbS. Patients treated with voxelotor showed increased levels of hemoglobin and other measures of hematological improvement including reduction in hemolysis and the percentage of sickled cells. Clinical trials (hereand here) have shown that voxelotor is effective.
The only other current pharmacological treatment of sickle cell disease is hydroxyurea, which induces the expression of fetal hemoglobin. This relieves some symptoms of sickle cell disease but is not a perfect solution. Fetal hemoglobin expression normally ceases after birth and does not have optimum oxygen-binding properties outside the uterus. On the other hand, voxelotor directly targets the cause of sickle cell disease and is well tolerated at high doses. The drug can also be taken safely by children, which could lessen age-related progression of sickle cell disease. Is voxelotor (OXBRYTA to Big Pharma) the answer to sickle cell disease? Not yet, but so far the drug is safe and outcomes are promising. Proof of principle is strong. Other HbS polymerization inhibitors are under development, and one or more may work better than voxelotor. For now, this drug is the only treatment that directly attacks the proximal cause of sickle cell disease.
The two biomedical developments described here, AAV-5-mediated gene therapy for hemophilia and voxelotor for sickle cell disease, are definitive triumphs of modern molecular biology and biochemistry. Both also show that the rational development of novel therapies for these two devastating diseases has been incremental, not to mention slow. From gene to effective therapy has taken about 40 years for HA and HB. Linus Pauling and his team described the polymerization of HbS in 1947 and noted that this was probably the cause of sickled cells and therefore the disease. The first inklings of a successful inhibitor of HbS polymerization appeared 70 years after the original hypothesis was put forward. One does wonder why it took so long to look for the obvious.
Now that we have these therapies, how widespread will be their use? Amit C. Nathwani in the previously linked paper at Figure 1 is frank. His conclusion includes a section on Affordability of AAV gene therapy:
It is likely that gene therapy will command a high price, at least initially, in order to recoup the development cost. However, gene therapy offers the advantage of continuous endogenous expression of clotting factor, which will eliminate breakthrough bleeding and microhemorrhages, thereby reducing comorbidities and the need for frequent medical interventions while improving quality of life. Thus, gene therapy has the potential to yield significant savings for the health care system and society in general but may still prove to be unaffordable for patients living in developing or emerging economies.
No doubt. But in those parts of the Global North that have universal healthcare, for the time being, instead of something called “access to health care,” the data are clear regarding the use of voxelotor for the treatment of sickle cell disease. Last month from France:
In 6100 modeled patients with SCD treated with voxelotor, the model projected the number of deaths to decrease by 39.4%, with an increase of 1.8% in life-years gained. The model also projected life expectancy to increase by 15.8%, and incident cases of stroke, pulmonary hypertension, and chronic kidney disease to decrease by 19.8%, 24.5%, and 25.1%, respectively. The model suggests that improving Hb using a treatment such as voxelotor may have a positive public health impact by reducing the burden of SCD for patients and the healthcare system.
These are incremental but significant improvements. Other polymerization inhibitors that follow this research may produce better results [9], as has been demonstrated for other genetic diseases such as cystic fibrosis (planned for next time). For now, the price of voxelotor that “someone” is paying ranges from $8,000 to $14,000 per month in the United States. I suppose this cost would not have been a problem for Nicholas II and Alexandra after the birth of the Tsarevich in 1904. For most of us the very notion is both outrageous and frightening.
Biomedical science has answers. Biomedicine gets in the way with too many extraneous questions. Therefore, Two Cheers for Biomedical Science. We can change that to three cheers. Our choice.
Notes
[1] Superfluous high school biology reminder: Humans have 23 pairs of chromosomes. 22 are autosomes and one pair are the sex chromosomes X and Y. Females are XX and males are XY. An uppercase letter is the symbol for the normal gene or the chromosome carrying the normal gene, while lower case is the symbol for the mutant gene or chromosome. The mutant can be less active than the normal version or completely absent or otherwise defective. 50% of the normal amount of F8 or F9 in the carrier female is more than sufficient for normal hemostasis.
[2] This pedigree is Lesson #1 that marrying your cousin is a bad idea. The Spanish Hapsburgs provide Lesson #2: From that Abstract, “It is speculated that the simultaneous occurrence in Charles II (F = 0.254) of two different genetic disorders: combined pituitary hormone deficiency and distal renal tubular acidosis, determined by recessive alleles at two unlinked loci, could explain most of the complex clinical profile of this king, including his impotence/infertility which in last instance led to the extinction of the dynasty.”
[3] OMIM was started by Victor A. McKusick, who edited entries in OMIM, often those he originated, until his death at the age of 86. OMIM is no cost to the user and up to date. I have donated so you do not have to if asked.
[4] I was there and began clone my first gene in 1982; we produced the clone in 1986. Four years in 1982 has become overnight in 2023, because most human cDNA clones are available for purchase. Gene therapy is useful but has not lived up to the early and unrealistic hype for a variety of reasons. The route from “in theory” to “in the clinic” (often referred to as “bench-to-bedside”) is not particularly long in conception, but it can be unusually crooked and often full of dead ends in execution.
[5] This was also before PCR made the amplification of the target gene very simple, when DNA sequencing was manual – a few hundred base pairs at a time, and expression of recombinant proteins was a black art. A usable human genome sequence, which is the lexicon that makes genetic medicine possible, also still lay about 20 years into the future.
[6] Given that sickle cell disease is debilitating, the persistence of mutant Hemoglobin S (HbS) was something of a mystery until it was determined that people with sickle cell trait (one copy of HbS beta-subunit and one copy of normal Hb-beta) are resistant to malaria. The malaria parasite does not survive well in red blood cells carrying the HbS protein. HbS is most common in equatorial Africa and the Mediterranean basin, where malaria is/was endemic. An accessible review of this is here.
[7] Linus Pauling was awarded his Nobel Prize in Chemistry in 1954 for his work on fundamentals of the chemical bond. Every chemist and biochemist should have a copy of The Nature of the Chemical Bond on the shelf; mine is staring back at me while I type this sentence. Pauling is also responsible for the alpha-helix, which is the primary structural motif in the protein chains of hemoglobin (a tetramer of two alpha and two beta subunits, or two alpha and two gamma subunits). Linus Pauling was awarded the Nobel Peace Prize in 1962 for his opposition to the nuclear arms race. S.J. Singer wrote, along with Garth Nicholson, the seminal paper (paywall) on the nature of biological membranes in 1972 that opened the door for future studies of membrane biology (another paywall).
[8] Hemoglobin polymers in sickle cells polarize light as it passes through the cells, leading to “birefringence” that can be measured with a simple device (polarimeter) that detects the transmission of polarized light through a sample.
[9] The other treatment of sickle cell disease that will work in principle is hematopoietic cell transplantation. Stem cell transplants are clearly indicated for various leukemias, but they also remain a dangerous, last resort, near shot in the dark under most conditions, which lack the perfect donor. Although “we have a pill for that” has become too common in modern medicine, a pill for sickle cell disease is much to be preferred to the destruction and rebuilding of the bone marrow. A better pill would be better yet.
