

Yves here. KLG unpacks a recent overview paper on SARS-CoV-2 mechanisms. Although this discussion is, of necessity, somewhat technical, Covid isn’t going away any time soon. So making an investment in understanding it better should help, if nothing else in discerning whether various commentators know what they are talking about.
By KLG, who has held research and academic positions in three US medical schools since 1995 and is currently Professor of Biochemistry and Associate Dean. He has performed and directed research on protein structure, function, and evolution; cell adhesion and motility; the mechanism of viral fusion proteins; and assembly of the vertebrate heart. He has served on national review panels of both public and private funding agencies, and his research and that of his students has been funded by the American Heart Association, American Cancer Society, and National Institutes of Health
We are 40 months into the COVID-19 pandemic, and we still do not have a good handle on how SARS-CoV-2 causes disease. Yes, the clinical correlates of COVID-19 are well described if somewhat less well understood, especially the nature of long COVID. Clinical management of disease has improved, and the pandemic has been declared over by the President of the United States. Nevertheless, several hundred Americans die every day of COVID-19, and this is undoubtedly an underestimate. Various interested parties argue about the significance of excess mortality over the past several years, but there is little real doubt about the cause.
Even so, the molecular and cellular causes of COVID-19 disease have not been described well enough for virologists and clinicians to have a sure grasp of what they, and we, are dealing with. Given that the response of the scientific community (>340,000 papers in PubMed with “COVID” as the query as of 11 March 2023) has been so large, this seems a bit strange (1). Nevertheless, a paper published in Nature Cell Biology on 9 March 2023 has made what is likely to be real progress: SARS-CoV-2 infection induces DNA damage, through CHK1 degradation and impaired 53BP1 recruitment, and cellular senescence (Open Access).
Although that is a very technical title, the data are presented very clearly and completely. The objective of this post is to guide the reader through representative experiments and their interpretation. I believe this is important because the explosion of COVID-19 literature has been so overwhelming. A lot has been published, and some of it has been hurried. More of it has been more “sciency” or “scientistic” than truly scientific. Fashion in biomedical science is certainly a thing (a subject for another time). This paper is one of the best I have read on the molecular and cellular derangements associated with COVID-19.
All that is really needed to appreciate the results in this paper is some high school biology:
- According to the Central Dogma of Molecular Biology, DNA makes RNA makes protein, and that proteins are the working parts of the process and virtually all other cellular functions. DNA is a double helix of A-T and G-C base pairs.
- All cells have mechanisms to ensure that DNA damage is repaired before cell division is completed, and that when maintenance of genome integrity is disrupted normal cellular functions will fail and this failure results in disease.
- Viruses are “organisms” that replicate only when they infect host cells and viral proteins allow the virus to hijack normal cellular processes to produce new virus particles. A pathogenic virus causes disease as a consequence of its life cycle or the activity of one or more of its proteins. Common viral diseases, historical and current, include polio, smallpox, AIDS, various conditions caused by herpes viruses, cancer (e.g., cervical and head/neck cancer caused by HPV, human papilloma virus), and COVID-19. Virus genetic material can be RNA or DNA.
Understanding this paper requires some guidance on the interpretation of the data. This is really no more difficult than understanding why Silicon Valley Bank failed last week. I am sure by the time this post appears we will both know more about the causes and consequences and responses of that untoward event. (As it turns out on Monday evening, 13 March 2023: “Nothing to see here, move along” seems to be a common trope among the more self-interested parties). The COVID-19 biology described here is certainly no more difficult to understand than “private equity,” derivatives, and credit default swaps, which are also maledictions that afflict us all in one way or another.
The coronavirus SARS-CoV-2 has a small RNA genome of 30 kb (30,000 RNA nucleotides in one single RNA molecule (2). This genome encodes 26 proteins, including 16 non-structural proteins (NSPs), 4 structural proteinssuch as the nucleocapsid (N) protein and 6 accessory proteins; the Spike protein of SARS-CoV-2 is the antigen for the COVID-19 mRNA vaccines. The DNA Damage Response (DDR) is a network of essential pathways that sense DNA lesions, signal their presence, and coordinate repair of the DNA before cell division can proceed without the mutations that would otherwise result if genome stability were not maintained. These DNA lesions include single-strand and double-strand breaks (SSBs and DSBs) in DNA, which are detected by various proteins in the cell. Some research has been done on DNA viruses and DDR, but less is known about RNA viruses. While it has been suggested that SARS-CoV-2 infection affects host cell DDR machinery, not much was known about DDR and RNA viruses before this paper was published last week (9 March 2023). Two proteins encoded in the SARS-CoV-2 genome are the culprits in interference with the DDR. They are ORF6 (“open reading frame” designates a protein sequence of previously unknown function that has been identified in a DNA or RNA sequence) and NSP13.
Paraphrased from the Introduction:
SARS-CoV-2 infection causes DNA damage and activation of an altered DDR. DNA damage is the consequence of the degradation of the cellular protein CHK1 by ORF6 and NSP13…Depletion of CHK1 leads to a shortage of the building blocks of DNA (the four building blocks – dNTPs – of DNA: dATP, dGTP, dCTP, dTTP; or A-T and G-C base pairs of the DNA double helix). This deficiency results in impaired cell division, DNA damage accumulation, DDR activation, induction of inflammatory pathways and cellular senescence. Supplementation with precursors of dNTPs is sufficient to counteract this cascade of events by allowing DNA replication to proceed. SARS-CoV-2 N-protein also interferes with DNA repair. In addition to the cultured human cells used as an experimental model, these events also occur in mice infected by SARS-CoV-2 and in patients with COVID-19.
Thus, this paper has identified several molecular concomitants of inflammation and associated cellular pathologies well known to afflict COVID-19 patients. In what follows I will illustrate some of the what and the why of these conclusions. The data look like cuneiform tablets at first glance, but only a few keys are necessary to understand them. This will be useful to interested readers who want to go deeper into the results of this paper, which is freely available and well worth the effort if productive rabbit holes are alluring. In my view, this is an exemplary scientific publication and, under the circumstances of COVID uncertainty, much needed. This primer will also make it possible to go beyond the Abstract and Conclusions of any other COVID-19 paper that concentrates on the molecular and cell biology of COVID-19. The devil is in the details, which have been devilishly lacking in too much of the extensive COVID-19 literature.
SARS-CoV-2 infection activates the DDR cascade and leads to CHK1 deficiency, which interferes with DDR (Figure 1a, paper). Controls are in the first of the four lanes, i.e., no SARS-CoV-2. This represents the basal or normal state of these cells [Huh7, a human liver cell line (3) naturally permissive for SARS-CoV-2 infection)] in the absence of SARS-CoV-2. Experimental samples were taken at 1, 24, and 48 hours post-SARS-CoV-2 infection. The top panel in the excerpt below (F-1-a) shows DNA-PK, which is a master regulator of DDR that must be activated by having a phosphoryl group added to it, denoted by pDNA-PK(S2056) (4). These two panels show that DNA-PK levels remain the same from time zero through 48 hours of infection but that the active phosphorylated version, i.e., pDNA-PK(S2056), has increased (red arrows). At the same time, the inverse is seen with CHK1 (red arrows). Both the protein and its active phosphorylated version are severely depleted at 48 hours, with this result: No CHK1, no downstream functional DDR despite the activation of the master regulator DNA-PK. Similar results are seen in the right panels with treatment by hydroxyurea or ionizing radiation, both of which disrupt DNA replication, although the response with CHK1 is not as strong as with SARS-CoV-2 infection. The bottom panel shows that SARS-CoV-2 N-protein increased during the experimental time course due to a productive infection with the virus. Taken together, the data presented here and in other experiments described are basically as good as they get.
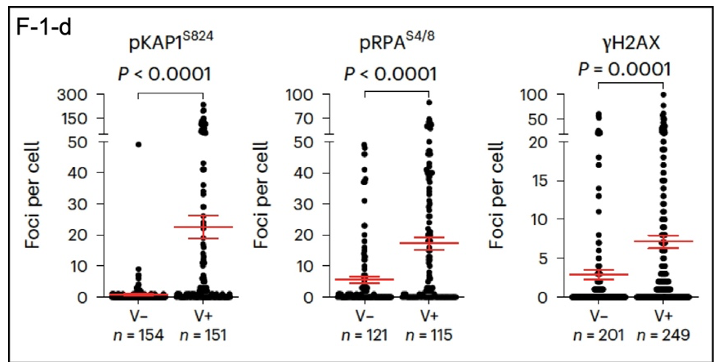
What happens in individual cells? DDR markers can be identified using imaging of proteins tagged with fluorescent reporters in infected cells; DAPI stains DNA (Figure 1c; in the paper). The fluorescence images are best viewed enlarged in the original, so I will concentrate here on quantitative data extracted from the images. These results are presented below (F-1-d). Infection with SARS-CoV-2 (V+) increases the number of DDR foci in infected cells, as identified by phosphorylated DDR proteins pKAP1, pRPA, and gamma-H2AX, with significantly more positives in virus-infected cells. Another significant result shown here is the variability of the effects of the virus on individual cells. This may be reflected in the variabilityof the course of disease in individual patients.
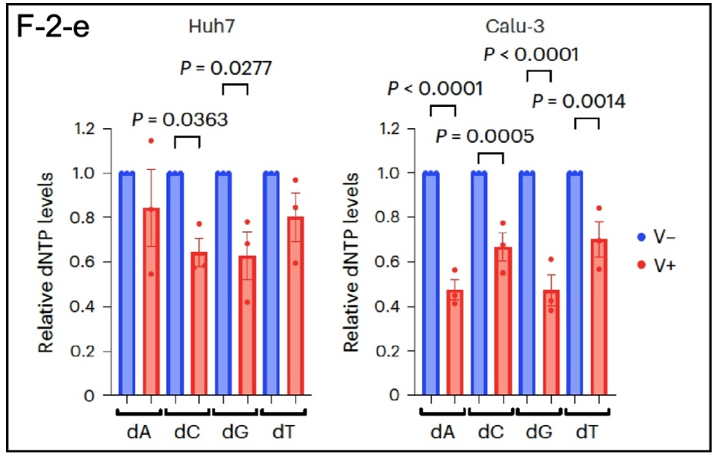
As shown in F-2-e, SARS-CoV-2 also reduces RRM2 levels (5), which leads to a shortage of the precursors for DNA synthesis (dATP, dCTP, dGTP, and dTTP), which are reduced by as much as 50% in Huh7 cells (liver) and Calu-3 cells (lung adenocarcinoma). This perturbation interfered with normal cell division and made it difficult for cells to proliferate. In the organism this leads to DNA damage and the secretion of inflammatory cytokines. Systemic inflammation is a major consequence of SARS-CoV-2 infection.
Which SARS-CoV-2 proteins are the culprits in these cellular perturbations? The authors of this paper individually expressed 24 of the 26 viral proteins in Huh7 cells using conventional techniques. Expression of both ORF6 and NSP13 reduced CHK1 and RRM2 protein levels by
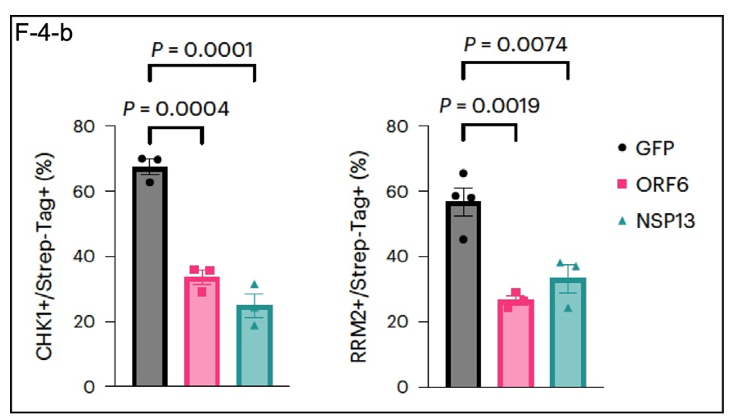
~50% (F-4-b). GFP (described here) as an exogenous protein control, so this result is not likely to be due to a general effect on protein expression by the viral proteins ORF6 and NSP13.
As mentioned in endnote (3), this kind of research on cultured cells is useful, but whether the cells are derived from tumors or immortalized by transformation with a tumor gene or cultured as normal primary lung or liver cells from the organism, they do not recapitulate the organism. Figure 7 in the paper (excerpt not shown here) shows that SARS-CoV-2 infection causes DNA damage in the lungs of mice expressing the human ACE2 protein along with expression patterns of DDR proteins expected from the experiments on cultured cells. ACE2 is the “receptor” (6) for SARS-CoV-2. As expected, CHK1 and RRM2 levels decreased in these “humanized” mouse lungs challenged with SARS-CoV-2.
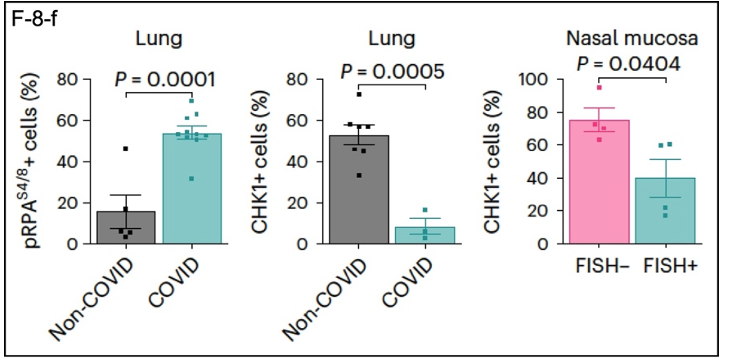
Data from samples of the lungs of COVID-19 patients (F-8-f) are also consistent with the overarching theme of this research. DDR-related proteins show expression patterns that complement those in the other experiments. pRPA is elevated in COVID lung tissue compared to non-COVID tissue, while CHK1 is depleted. A similar result was obtained for CHK1 using nasal mucosal tissue, where SARS-CoV-2 enters the body.
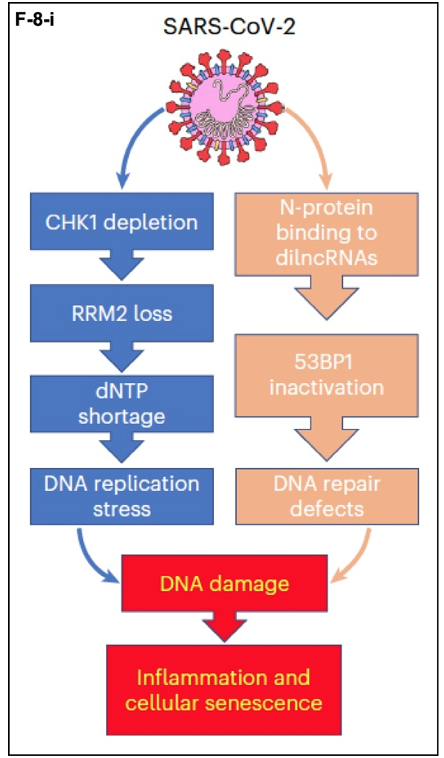
The results of this research can be summarized as follows in (F-8-i). SARS-CoV-2 infects target cells and exerts its pathological effects through two parallel pathways required for maintaining genome integrity. In the mechanism described here, CHK1 depletion leads to depletion of RRM2. The resulting dNTP shortage leads to DNA replication stress and DNA damage. In the parallel pathway, for which the data are also strong, the SARS-CoV-2 N-protein binds to damage-induced, long non-coding RNAs (dilncRNAs). These RNA molecules (7) nucleate DDR complexes at sites where they are required. The viral N-protein disrupts this process and leads to DNA repair defects. The end results are senescence of the infected cells, i.e., they get old and die before their time, and inflammation caused by the death of these cells.
Why in my view is this an exemplary paper? From the ground up the hypothesis is well conceived: SARS-CoV-2 infection dysregulates the DNA Damage Response in individual cells. This is demonstrated in multiple, complementary experiments, only a representative few explained here. The paper then shows that effects on DDR are also present in the animal model represented by the mouse expressing human ACE2. Finally, examination of samples from COVID-19 patients shows that SARS-CoV-2 affects pRPA and CHK1 expression as would be expected based on extensive analyses using cultured cells and the mouse model. From the conclusion to the paper:
Altogether, our results indicate that SARS-CoV-2-induced DNA damage triggers a cell-intrinsic pro-inflammatory programme that, in concert with the immune response, fuels the strong inflammatory response observed in patients with COVID-19. The observed ageing phenotypes recently reported in patients with severe COVID-19 are consistent with our observations. Finally, by proposing a mechanism for the generation of DNA damage and the activation of DDR pathways and of a pro-inflammatory programme, we provide a model to improve our understanding of SARS-CoV-2-induced cellular senescence. In this regard, it will also be interesting to determine if persistent DNA damage and DDR activation, features of cellular senescence, following SARS-CoV-2 infection, contribute to the chronic manifestations of the pathology known as long COVID. (emphasis added)
I see nothing to add to this (8). The results explain much of what is observed in COVID-19 patients, and this research will be useful in understanding long COVID. How this knowledge might lead to effective treatments of COVID-19 is not immediately clear, but a thorough understanding of the molecular and cellular derangements caused by COVID-19 will be essential. As shown in this research, the damage done by SARS-CoV-2 probably resides deep in the cell and can be severe and long lasting for that reason. In the meantime, wear your mask (N95 or better)!
Notes
(1) Within a relatively short time 40 years ago it became clear that HIV causes disease by reducing the population of T-cells of the immune system to levels that prevented the immune system from working properly. This is why AIDS is associated with opportunistic infections (e.g., cytomegalovirus, Pneumocystis jerovicii pneumonia) and other AIDS-associated diseases such as Kaposi sarcoma.
(2) How small is 30,000 base pairs? The coding regions of the two alternative forms of “my favorite protein,” which are on different chromosomes, are ~7,600 base pairs that produce proteins of 2,541 and 2,542 amino acids, respectively. The gene of one version is 27,000 base pairs long when the noncoding introns of the gene are included. The other gene is 300,000 base pairs long, or about 10 times the size of the SARS-CoV-2 genome.
(3) A “cell line” is a term of art denoting a population of immortal cells, i.e., those that do not eventually stop dividing in their “old age” (senescence) in culture; Huh7 cells are immortal cells from a liver tumor. Cell lines are essential for cell biology, but cellular results must be extended to other experimental models for them to be a valid biological result that can be extended to clinical medicine. This has been done in the research described here, which included experiments in mice infected with SARS-CoV-2 and analysis of tissues from human COVID-19 patients. The latter confirmed and extended the results obtained with cultured cells.
(4) pDNA-PK(S2056) = phospho-DNA-PK on the 2056th amino acid in the protein, which is the amino acid serine. Similar designations apply to the other proteins analyzed. These proteins have been separated by size (the 450 kDa marker = a molecular weight of 450,000), transferred to a permanent matrix and identified using antibodies specific for each protein. Immunoblotting is described here if you want do go down that rabbit hole.
(5) RRM2 is a subunit of the essential enzyme ribonucleotide reductase, which converts ribose-based nucleotides (RNA building blocks) to deoxyribose-based nucleotides (dNTPs, DNA building blocks). Without enough dNTPs available DNA synthesis is impaired and affected cells get sick.
(6) ACE2 is usually called the receptor for SARS-CoV-2. “Receptor” has a specific meaning in cell biology. T-cell receptors mediate immune responses. When insulin binds to the insulin receptor, a series of events leads to the clearing of excess glucose from the circulation after a meal. Both of these are normal homeostatic functions in the organism, one necessary for normal immune function and the other necessary for energy metabolism. In my view, ACE2 should more properly be called the binding protein that allows SARS-CoV-2 to enter human cells and cause disease. That the virus binds to ACE2 is an unfortunate evolutionary accident, not unlike CD4, which normally acts as a co-receptor with the T-cell receptor but also binds to HIV and facilitates viral entry into CD4+ cells. AIDS is the result.
(7) Long non-coding RNAs are relatively new to the RNA world of molecular cell biology. Their existence was a surprise, and they are involved in the regulation of gene expression and other processes such as the DNA Damage Response (DDR).
(8) I have at times expressed reservations about a biology paper with 29 authors. This research, however, took that many working scientists to complete in a timely fashion. The project was very large and multifarious. The paper was also under review for 13 months, from December 2021 until January 2023, during which a number of results must have been clarified or confirmed at the behest of the reviewers and editors. Only one author of 29 revealed any possible competing interests, which seem minor in the larger context of the research. The paper is also published in a legacy journal, which might be defined as one that predates the rise, and near dominance, of online “journals.” Nature Cell Biology (1999) qualifies. It has also been very strong in the cell biology community for almost 25 years. But that is not a guarantee. The Lancet (1823) published Andrew Wakefield’s paper on the (nonexistent) relationship between the childhood MMR vaccine and autism. The New England Journal of Medicine (1812) published the paper that confirmed the Pfizer/BioNTech COVID mRNA vaccine is safe and effective.
